La comunicación verbal como base para la interrelación
Dra.
María Teresa Ferrando Lucas Médico
neuropediatra
Universidad Complutense de Madrid
Centro de Rehabilitaci?n del Lenguaje
LA
COMUNICACIÓN VERBAL COMO BASE PARA LA INTERRELACIÓN
INTRODUCCIÓN
La
voluntad que subyace en el informe de la UNESCO concerniente
a la educación en el siglo XXI conduce al completo
desarrollo del ser humano.
El aprendizaje tiene un requisito previo: la adquisición
del lenguaje. La comunicación constituye la base
de la convivencia: el poder expresar deseos, sentimientos,
creaciones, costumbres, ideas y el comprender lo que
nos transmiten otros seres humanos requiere que seamos
capaces de dominar un código a través
del cual podamos comunicarnos. Puede ser argumentado
que existen muchas formas de lenguaje, en tanto que
son numerosos los mensajes que podemos recibir del entorno
y no solo del ser humano. Los animales nos transmiten
mensajes mediante sonidos y gestos, que somos capaces
de interpretar. Incluso podemos interpretar mensajes
transmitidos por el mundo vegetal en razón de
variaciones de forma y color. Podrá ser argüido
la importancia del lenguaje gestual entre personas.
Sin embargo, ninguna de estas formas de expresión
posee la riqueza de la función mas fina del cerebro
humano, base del pensamiento y base de la comunicación:
el lenguaje oral y su codificación en forma de
lenguaje escrito.
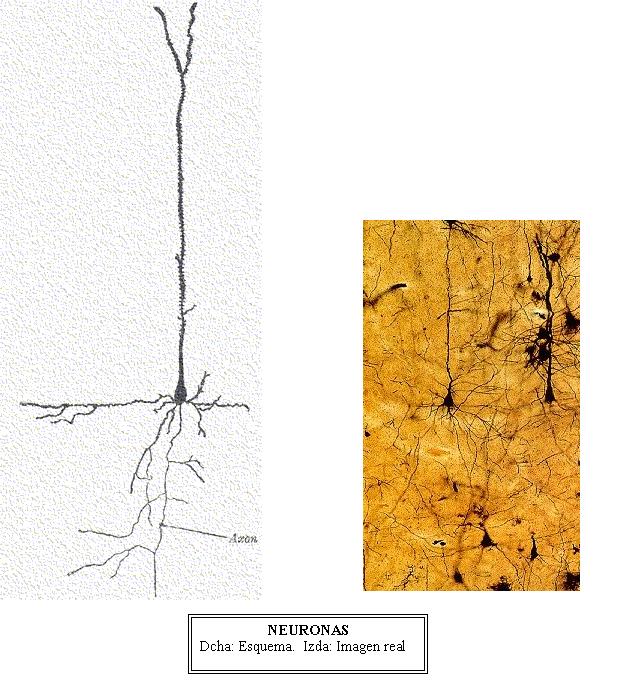
El
cerebro humano está genéticamente programado
para asentar el lenguaje oral. Alrededor del día
100 de la gestación, todas las neuronas que vamos
a poseer a lo largo de nuestra vida han terminado de
formarse en una zona profunda de este cerebro en formación
denominada matriz germinal ; a partir de este momento
las neuronas emigran hasta ocupar su lugar definitivo
en la corteza cerebral.
Este
proceso de migración neuronal se ve precedido
por células guía, que abren caminos por
los que las neuronas viajaran y se irán colocando
en capas formando el cortex cerebral. Una vez cumplida
su misión las células guía involucionan
hasta desaparecer.
El
fin último del sistema nervioso es procesar y
transmitir información. Para conseguirlo las
neuronas deben establecer conexiones. La maduración
del sistema nervioso central queda definida por dos
fenómenos: Histogénesis, durante la que
se forman las células nerviosas y adoptan una
disposición determinada y hodogénesis,
que permite la conexión entre células
por crecimiento de las sinapsis. La hodogénesis
permite que las células nerviosas se comuniquen
entre sí y se transmitan la información
procesada y almacenada a lo largo del circuito. La histogénesis
tiene lugar durante la gestación con participación
primordial de los factores genéticos, mientras
que la hodogénesis es un proceso que se prolonga
a lo largo de la vida extrauterina y en ella intervienes
factores externos como la nutrición y el aprendizaje:
No podemos influir en el número de neuronas,
pero sí en la riqueza y multiplicación
de las conexiones que establecen entre ellas (1, 2).
Esto implica una seria responsabilidad puesto que tanto
la educación como la alimentación de los
seres humanos depende de otros seres humanos y en último
término la correcta maduración del sistema
nervioso va a tener una dependencia del entorno sociocultural
importante y definitiva, que va a actuar ya desde el
momento de la concepción puesto que el tipo de
vida y alimentación de la madre van a repercutir
en el correcto desarrollo del feto. La carga genética
patológica que pueda condicionar desviaciones
de un normal desarrollo no podemos modificarla, pero
si podemos influir de modo positivo sobre la maduración
del sistema nervioso mediante la educación; el
aprendizaje influye en la riqueza de conexiones (sinapsis)
que las neuronas establecen entre ellas y este proceso
no termina nunca ya que somos capaces, en condiciones
normales, de seguir aprendiendo, aunque lentifiquemos
el ritmo, a lo largo de toda nuestra vida.
Sobre
el cerebro maduro, en cuanto al número de neuronas,
empiezan a asentarse las funciones del lenguaje, como
resultado de un proceso de aprendizaje, que se extiende
a lo largo de varios años: La fonología
requiere de los cuatro primeros años de vida para
terminar su implantación y la sintaxis de los seis
primeros años. Las nuevas tecnologías de
imagen cerebral funcional nos están llevando a
modificar muchos de nuestros actuales conceptos sobre
lenguaje y aprendizaje pero también a confirmar
los conocimientos previos sobre las áreas cerebrales
implicadas en estas funciones. Las imágenes que
siguen nos ayudarán a recordar de modo esquemático
las zonas cerebrales responsables del lenguaje.
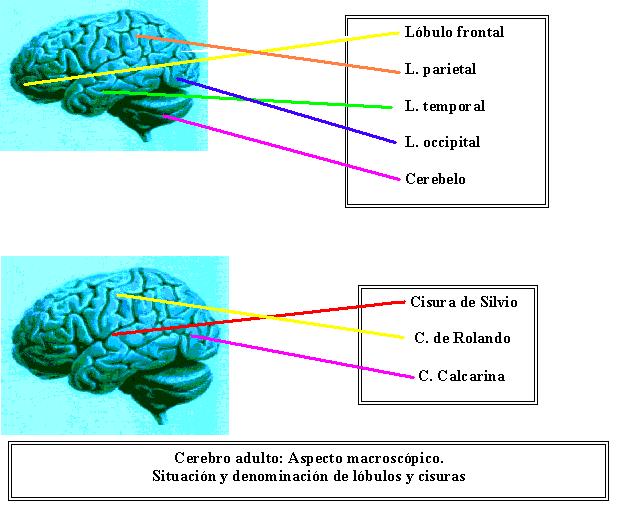
ÁREAS
DEL LENGUAJE
Areas
cerebrales del lenguaje
Para
un gran porcentaje de la población (87%), el
lenguaje se asienta en una zona amplia del hemisferio
izquierdo; mientras que para un pequeño porcentaje
(8%) se localiza en el hemisferio derecho y el resto
(5%), lo distribuye en ambos hemisferios, sin una clara
definición. La lateralización del lenguaje
llevó a pensar que el hemisferio izquierdo era
el dominante, aunque se ha comprobado que la lateralización
del lenguaje no siempre coincide con la lateralidad
manual (2). La tendencia entre los especialista, hoy
día, es a dejar de lado el concepto de hemisferio
dominante y a considerar que ambos hemisferios son preponderantes,
dependiendo de la función (Tabla 1).
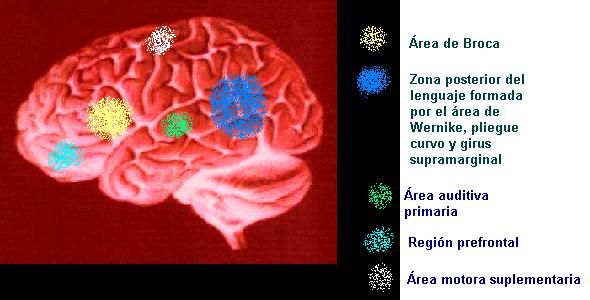
Areas
de la comprensión del lenguaje
Las
señales acústicas (fonemas, palabras)
son transformadas en el oído interno en señales
bioeléctricas, las cuales son conducidas por
el nervio acústico hasta el SNC, específicamente
al área auditiva primaria (gyrus de Heschl).
El área de la audición primaria se ubica
en la parte superior de los lóbulos temporales,
en la profundidad de la cisura de Silvio. Cuando la
señal llega a esta zona es cuando se interpreta
como un sonido. Sin embargo, para interpretarse como
parte del lenguaje, debe activarse el área de
Wernicke formada por el planum temporale y una parte
de la primera circunvolución temporal, en el
hemisferio izquierdo. A través del cuerpo calloso,
el área de Wernicke recibe información
de
las señales que provienen del gyrus de Heschl
derecho. La activación del área de Wernicke
permite reconocer los fonemas, como parte fundamental
del lenguaje. Para asignar a los fonemas categoría
simbólica, se han de activar otras dos zonas
del hemisferio izquierdo: el gyrus supramarginal para
el análisis morfosintáctico y el pliegue
curvo para el análisis semántico. El pliegue
curvo es también la zona donde se realiza la
correspondencia de significantes y significados, tanto
para la lengua oral como para la lengua escrita. Su
función de córtex terciario asociativo
plurisensorial comprende además la identificación
de gnosias visuales, táctiles y auditivas no
verbales.
Areas
de la expresión del lenguaje.
La
elaboración del habla necesita a su vez otros
procesos como la formulación y programación
motriz que finaliza en el acto fonoarticulatorio para
la producción del habla. El área implicada
en este acto es la parte inferior del lóbulo
frontal, por delante de la cisura de Rolando. En el
tercio posterior de la tercera circunvolución
frontal izquierda se encuentra el área de Broca
con dos partes importantes. Una anterior, denominada
pars triangularis cuya misión es formular
el esquema práxico del mensaje verbal. Otra posterior,
denominada pars opercularis, que da la orden
de emitir el movimiento adecuado para producir los fonemas
de las palabras.
Otras
zonas cerebrales implicadas en la expresión son
el área prefrontal, el área motora suplementaria
y los ganglios de la base.La prosodia depende del hemisferio
derecho.
Tabla 1 - SUBSTRATO
CORTICAL DEL LENGUAJE
HEMISFERIO IZQUIERDO
Fonología.
Sintaxis.
Semántica.
Acceso al léxico.
Adaptación del menaje formuladoa su
contenido semántico.
Conversión de signos gráficos
en estructuras sintácticas y semánticas.
HEMISFERIO DERECHO
Atención.
Orientación espacial.
Prosodia.
Adecuación del lenguaje al contexto.
Visomotor
AMBOS HEMISFERIOS
Iniciativa verbal.
Memoria verbal.
Tonalidad afectiva.
Identificación de signos gráficos.
Cuando
en el proceso de maduración de un niño,
el desarrollo del lenguaje no es adecuado, y no existen
motivos sociales ni culturales que lo justifiquen ,
debemos plantearnos las siguientes posibilidades:
La
primera que se trate de un retraso madurativo simple;
el apoyo logopédico suele ser suficiente.
La
segunda posibilidad es que estemos enfrentándonos
a un trastorno específico del desarrollo del
lenguaje, es decir a una disfasia. En su diagnóstico
y tratamiento es necesaria una actuación multidisciplinar
en la que logopedas, psicólogos, maestros,
pedagogos y médicos se ven implicados.
En tercer lugar debemos contemplar la posibilidad
de una patología neurológica, en la
que el trastorno del lenguaje constituiría
una manifestación mas, en ocasiones previa
al desarrollo completo de los síntomas de una
enfermedad concreta.
Llegados a este punto podemos
entender que el educador juegue un papel a la vez de
gran responsabilidad y de privilegio: de gran responsabilidad
en tanto que su cometido profesional tiene una repercusión
directa en la maduración del sistema nervioso.
De privilegio en cuanto a la capacidad de poder detectar
precozmente las desviaciones de la normalidad y dar
la voz de alarma para poner en marcha los mecanismos
diagnósticos y terapéuticos adecuados
a cada caso.
El educador se enfrentará a niños que
no comunicaran porque no saben o no pueden y esto ya
implica un fino poder de observación en el aula:
determinar si estamos ante un niño con autismo
o con un trastorno específico del desarrollo
del lenguaje puede llegar a ser una tarea muy difícil.
Por su frecuencia como causa de trastorno del desarrollo,
el cromosoma x frágil debe ser entidad conocida
por los educadores. La relación entre epilepsia
y trastornos de la comunicación y cognición
es un tema que suscita múltiples dudas tanto
en el entorno escolar como familiar. Estos van a ser
los puntos que van a ser abordados a lo largo de esta
ponencia.
RETRASO
INTELECTIVO: CROMOSOMA X FRÁGIL
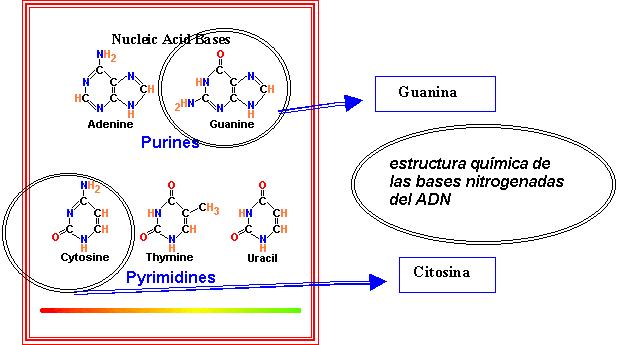
El síndrome X frágil
es una afección de origen genético cuya
manifestación clínica guía es el
retraso mental y cuya causa es la expansión de
la tripleta formada por las bases nitogenadas del ADN
citosina- guanina- guanina , debido a la inactivación
del gen FRM1 ó gen del retraso mental por X frágil
(3).
La frecuencia estimada, en todo el mundo, supone que
1 de cada 4000 varones y 1 de cada 6000 mujeres están
afectadas por la enfermedad. En el caso de portadores
se estima 1 cada 260 mujeres y 1 por 800 hombres. Estas
cifras extrapoladas a nuestro país pueden hacer
concluir que existirían alrededor de 10.000 afectados
y 100.000 portadores aunque en España no tenemos
un censo que contemple la totalidad de los casos.
Descrito por primera vez en 1943 por Martin y Bell,
el síndrome posee unas características
clínicas definidas por un fenotipo físico
y un fenotipo conductual, a los que se asocian otras
manifestaciones orgánicas (4). La correlación
con una alteración citogenética fue descrita
en 1969 por Lubs, quién empleando cultivos de
linfocitos en medio pobre en ácido fólico
apreció una fragilidad del cromosoma X en el
locus q27.3. En 1991 el defecto molecular consistente
en la alteración del gen FMR1, codificador de
la proteina FMRP, la cual se encuentra muy disminuida
o ausente en los individuos afectos, fue establecido
por tres grupos de investigadores: Oberle, Yu y Verker,
junto a sus respectivos equipos. La proteína
juega un papel fundamental en el desarrollo de diversos
tejidos, siendo su expresión mayor en testículo
y cerebro.
Considerado
como la primera causa de retraso mental hereditario,
el fenotipo físico viene definido por facies
y mandibula alargadas; orejas grandes y prominentes
y macroorquidismo. Este fenotipo clásico, sin
embargo, no esta establecido antes de la pubertad en
los niños afectos, por lo que la sospecha de
la enfermedad en edades anteriores debe establecerse
ante trastornos del desarrollo motor y del lenguaje
así como trastornos de conducta siendo el mas
precoz la hiperactividad y el mas grave el autismo.
Hipersensibilidad a los sonidos; dificultad de interacción
social; actitud de defensa ante situaciones nuevas;
movimientos estereotipados y mal contacto visual son
otras de las características que pueden presentar.
En las niñas no existe un fenotipo físico
tan definido como en el varón debiendo sospecharse
por los trastornos del desarrollo, conducta y dificultades
de aprendizaje, con cociente intelectivo bajo.
Otras alteraciones descritas son hiperlaxitud, por alteración
del tejido conjuntivo, otitis de repetición,
estrabismo y alteración cardiaca consistente
en prolapso de la válvula mitral. Han sido descritas
igualmente alteraciones morfológicas del sistema
nervioso central, puesto de manifiesto con Resonancia
Magnética, habiendose descrito aumento de los
ventriculos laterales e hipoplasia de vermis cerebeloso.
Existen igualmente publicaciones que hablan de una mayor
tendencia de estos niños a desarrollar epilepsia
fronto- temporal.
El único medio de asegurar el diagnóstico
es el estudio de genética molecular, analizando
la secuencia repetitiva CGG. Esto se efectúa
mediante un análisis de sangre, a partir del
que se obtiene ácido desoxirribonucleico (ADN)
y sobre el que se estudia la expansión de la
tripleta referida.
El
número de repeticiones, en la persona no afecta
oscila entre 6 y 50. Cuando su número se encuentra
entre 50 y 200 se habla de estado de premutación.
Esta es la situación del portador sano, pero
con riesgo de tener hijos afectados. Por encima de 200
repeticiones el gen se encuentra en estado de mutación
completa: es la situación de los pacientes que
desarrollan la enfermedad.
Debe
sospecharse siempre ante cualquier retraso del desarrollo
motor y/o del lenguaje de causa no conocida, así
como trastornos de conducta tales como la hiperactividad
y los trastornos de comunicación. Igualmente
debe siempre ser descartado ante retrasos intelectivos,
aún ligeros, y ante antecedentes familiares de
retraso intelectivo.
La causa más frecuente de diagnósticos
tardíos es infravalorar los trastornos del desarrollo
como síntoma, no teniendo en cuenta que en edades
tempranas es el único signo guía ya que
el fenotipo clásico no se va a encontrar en los
varones hasta la pubertad, y aún así hay
un 20% que pueden no presentarlo. Esta situación
se intensifica frente a las niñas que no van
a tener un fenotipo físico definido como los
varones.
Otra
fuente de error es el conformarse como método
diagnóstico con el cultivo de linfocitos en medio
pobre en ácido fólico ya que este solo
detecta los varones afectos, pero no las niñas,
en las que se dan muchos falsos negativos, ni a las
portadoras.
La mutación completa implica el desarrollo de
la enfermedad en la totalidad de los varones y en el
30 % de las mujeres.
En el caso de las premutaciones, depende del número
de repeticiones la posibilidad de pasar a mutación
completa y por tanto de tener hijos afectos. Existe
la posibilidad de determinar la situación, con
el fin de que el consejo genético sea adecuado.
Los casos de premutación pueden pasar a mutación
completa en la siguiente generación en el caso
de las mujeres, con lo que tendrán hijos afectos;
los varones con premutación tendrán hijas
sanas portadoras que tendrán hijos afectos (fenómeno
de anticipación genética). Existe igualmente
la posibilidad de efectuar el diagnóstico prenatal
cuando los padres son portadores.
El tratamiento contempla dos vertientes. Una
es la derivada de las líneas de investigación
encaminadas al tratamiento curativo de le enfermedad.
No es una posibilidad en el momento actual.
La
otra, que constituye el único medio eficaz en
el momento presente es el derivado de la detección
precoz de afectados, portadores y adecuado consejo genético.
La
posibilidad de simplificar el diagnóstico mediante
el estudio de cabellos, en los que se comprueba si existe
la proteina FMRP en el bulbo piloso abre nuevos caminos
al diagnóstico precoz; existen grupos de trabajo,
entre otros, en Rotterdam y en Zaragoza.
El
trabajo efectuado por el grupo de Sabadell, analizando
las características fenotípicas y conductuales
de estos niños, constituye una de las serie mas
amplias efectuadas hasta el momento en el mundo y la
mas importante en nuestro entorno. Es otra vertiente
del trabajo médico que contribuye a un mejor
conocimiento y por tanto prevención del Síndrome.
En la actualidad no existe tratamiento curativo. Se
utilizan tratamientos para algunos de los síntomas,
así metilfenidato para la hiperactividad, anticonvulsivos
si el niño padece epilepsia. Han sido propuestos
tratamientos con ácido fólico, con resultados
poco esperanzadores y existe una línea de trabajo
sobre el tratamiento con carnitina que en nuestro país
esta siendo desarrollado en la Universidad de Zaragoza,
dentro de un programa multicéntrico en el que
participan varios países de la Unión Europea.
El tratamiento fundamental es el psicológico
y pedagógico, con apoyo logopédico y escolarización
y posterior formación laboral acorde a sus limitaciones.
La
curación a corto plazo no es una posibilidad
real. La clave se encuentra en la terapia génica
y en ese sentido trabajan los diferentes grupos de investigación.
Se trabaja con ratones en los que se ha provocado la
mutación del gen FMR1 de modo que presentan la
expansión CGG como los pacientes X frágil.
Introducir el gen defectuoso, introducir la proteina
que falta e invertir la metilación que inactiva
el gen se cuentan entre las principales líneas
de investigación.
La creación de equipos pluriprofesionales (psicólogos,
pedagogos, médicos, trabajadores sociales) debería
ser otra de las metas de la Administración, así
como la integración social y laboral de todos
los pacientes.
EN
CONCLUSIÓN, el síndrome x frágil
debe ser diagnosticado precozmente por las implicaciones
familiares, pedagógicas, sociales y médicas
que conlleva. Es un error el descartarlo unicamente
frente a casos de retraso intelectivo y/o fenotipos
definidos. Los trastornos del lenguaje, conducta, comunicación
y aprendizaje deben constituir un punto de alarma y
hacernos pensar en esta patología.
EPILEPSIA,
LENGUAJE Y COGNICIÓN
El
concepto de epilepsia nos enfrenta a la definición
de un trastorno paroxístico que se manifiesta
en forma de crisis clínicas de cuya semiología,
etiología y correlato electroencefalográfico
va a depender la clasificación, tratamiento y
pronóstico de los diferentes síndromes
epilépticos.
La
repercusión de la epilepsia sobre la cognición
constituye un polo de gran interés y se postula
la posibilidad que los niños con epilepsia puedan
tener un menor rendimiento intelectual debido a daño
cerebral como consecuencia fisiopatológica de
las descargas epileptiformes (5-9). Al considerar la
relación de epilepsia y afectación del
lenguaje, dos entidades son a retener: la afasia epiléptica
ó síndrome de Landau y Kleffner (SLK)
(10- 14) y la epilepsia con punta- onda continua durante
el sueño (EPOCS)(14). El trastorno neuropsicológico
es la clínica mas dramática y de pronóstico
mas incierto en ambas. La epilepsia parcial benigna
atípica (EPBA) comparte con las dos anteriores
el trazado electroencefalográfico que adquiere
morfología de punta- onda durante el sueño
n- REM; aunque en la publicación inicial de Aicardí
y Chevrie (15) el estado mental de los pacientes es
descrito como normal, posteriormente se ha aceptado
la presencia de trastornos neuropsicológicos,
y las tres entidades (SLK, EPOCS, EPBA) constituirían
un continuum de la misma patología, que sería
capaz de manifestarse, en función de la edad
de comienzo, con diferente gravedad y pronóstico,
siendo la mas severa la afasia epiléptica y la
de mejor pronóstico la EPBA. (16, 17).
Que las descargas interictales subclínicas, tienen
consecuencias negativas sobre el lenguaje, conducta
y aprendizaje es un concepto que va siendo aceptado
a medida que se ve documentado en la literatura; han
sido apreciados en diversos tipos de epilepsia incluso
en aquellas consideradas de buen pronóstico tal
como la epilepsia rolándica (18, 19).
Descargas
electroencefalográficas epileptiformes sin correlato
clínico aparente han sido descritas en niños
autistas y disfásicos, así como una mayor
tendencia de estos pacientes a padecer epilepsia ( 20,
21). El significado de dicho hallazgo y su relación
con el trastorno neuropsicológico (TNP) es motivo
de gran controversia; si además del apoyo logopédico,
psicológico y pedagógico estos pequeños
deben recibir tratamiento con fármacos antiepilépticos
es motivo, si cabe, de mayor discusión. Es muy
clásica la afirmación que la alteración
aislada del electroencefalograma en niños normales
no debe tratarse. Ahora bien, ?por qué consideramos
normal a un niño cuya alteración del lenguaje
y la conducta es de tal entidad que le impide la adecuada
relación con el entorno y un correcto seguimiento
escolar?.
El
delimitar si el trazado e.e.g. patológico en
niños con trastorno severo del lenguaje tiene
un significado epiléptico o bien constituye un
marcador de mal pronóstico en una disfasia adquiere
interés prioritario frente a la actitud terapéutica
: El apoyo logopédico/psicológico/pedagógico
es imprescindible pero ?debe además establecerse
tratamiento farmacológico antiepileptico?. La
medicación por si misma puede parecer una contradicción;
ante un problema cognitivo y/o del lenguaje actuamos
con fármacos que poseen efectos colaterales negativos
sobre la cognición (22, 23). Sin embargo las
descargas infraclínicas interictales condicionan
alteraciones en el lenguaje la conducta y el aprendizaje
en pacientes con epilepsia (24- 25) así como
en patologías severas previas de la comunicación
(26), y una relación causa efecto parece existir
entre mejoría del eeg y la sintomatología
NP en pacientes con diferentes tipo de epilepsia (27-
28).
La
incidencia de trazados eeg alterados en niños
con disfasia es poco frecuente. En los casos recogidos
en la literatura los autores han efectuado su estudio
a lo largo de varios años y aún así
las series no son numerosas (30- 33) . Nuestra vivencia
del tema hasta el presente nos hace postular que efectivamente
no es alta; entre el año 96 y el momento actual
en tanto en c.r.l./ Madrid como en el Grupo de Neurología
Cognitiva del Hospital Sant Joan de Déu/ Barcelona
entre uno y tres niños de los que acuden anualmente
por TEDL presentan alteraciones eeg sin otro tipo de
manifestación clínica, y esta frecuencia
parece ser la misma entre los pacientes del Miami Hospital
Childrens (Prof. Pappazian- Comunicación personal)
?Por qué considerar que frente a este tipo de
trastorno estamos ante una epilepsia? ?Qué comparte
y en que es diferente la clínica de estos pequeños
cuando se les compara con niños en que el diagnóstico
de epilepsia no ofrece discusión?.
En
primer lugar son diferentes en cuanto a la fenomenología
motora. Quedan por tanto excluida todo tipo de epilepsia
que se manifieste con crisis clínicas detectables.
Quedarían también fuera de consideración
todas las que cursen con deterioro o regresión:
Estamos considerando trastornos específicos del
desarrollo del lenguaje y por tanto hay que ceñirse
a sus características clínicas (34).
El
SLK debe ser considerado con mucha calma; si revisamoss
cuidadosamente la bibliografía (35- 39): lo que
realmente lo define es el trastorno de la comunicación,
la afasia y la alteración eeg. No las crisis.
Están aceptados y descritos SLK sin crisis clínicas
demostrables. Tampoco excluye el padecimiento de la
entidad el no presentar agnosia auditiva (AA): la agnosia
auditiva es la manifestación mas severa de la
afasia, pero esta puede manifestarse también
en forma de alteración de la comprensión
en grados menos severos y aún mas, en forma de
trastorno expresivo dominando sobre el receptivo. El
trazado eeg es multifocal con tendencia a la difusión
intra e interhemisférica , que se activa de modo
importante durante el sueño n-REM pudiendo llegar
adquirir la morfología de punta- onda contínua,
pero el no alcanzar esta característica no excluye
la entidad.
Atendiendo
a la edad mas frecuente de comienzo, 3 a 7 años,
en un niño con un desarrollo previo del lenguaje
normal, la afasia no pasaría desapercibida, aún
en el caso que no presentase crisis clínicas,
pero ?qué pasaría a edades mas tiernas?.
El
caso mas joven de Deonna tiene 24 meses. Si seguimos
descendiendo en la edad, en los estadios del inicio
del lenguaje ?cómo detectamos la afasia?. Sin
embargo de afasia deberíamos hablar si un niño
pierde por causas patológicas su lenguaje: el
lenguaje que posea, es decir el correspondiente a lo
esperable atendiendo a su edad cronológica; si
está en la etapa de lalación o de primeros
bisílabos referenciales y ese es el lenguaje
que debe poseer y una noxa interfiere en la utilización
y desarrollo del mismo, de afasia deberíamos
clasificarlo. Pero esto es muy difícil de detectar.
Este niño algunos años después,
en función de su falta de adquisición
de lenguaje lo diagnosticaremos probablemente de disfasia.
Por
último también sabemos que el pronóstico
en cuanto a la recuperación del lenguaje, en
el SLK es peor a menor edad; es decir, cuando mas pequeño
es el paciente al inicio de la sintomatología
mas severa y persistente es la afasia. Las crisis, cuando
las hay, y el trazado eeg, tienden en todos los casos
a normalizarse, pero la afasia puede persistir.
Debemos
pues considerar que ante una disfasia que no evoluciona
y en la que encontramos un eeg paroxístico, que
tiende a mayor activación durante el sueño,
podemos estar ante un SLK de inicio tan precoz, que
la afasia haya pasado desapercibida.
La
relación entre descargas paroxísticas
y el trastorno del lenguaje no ha sido, hasta el presente,
bien esclarecida (40- 43).
La
relación entre epilepsia y trastorno del lenguaje
gira de modo reiterativo alrededor de síndromes
epilépticos bien aceptados La necesidad de encontrar
y probar que los paroxismos eeg tienen un efecto sobre
la cognición (44- 45) implica que dicha alteración
debe ser buscada y el buscarla implica un estudio laborioso
(46-49) que nos determine cual es realmente la situación
neuropsicológica de nuestros pacientes y su evolución
en función de las alteraciones paroxísticas
infraclínicas.
EN
CONCLUSIÓN: Las descargas paroxísticas
pueden estar traduciendo un daño cerebral tal
como se acepta en la epilepsia (50). La no constancia
de crisis clínicas no excluye que tal daño
se pueda estar produciendo. Es posible que el concepto
de epilepsia deba cambiar en el futuro (51); que algunos
trastornos cognitivos deban contemplarse como un tipo
de epilepsia. El tratar el eeg, un papel, sería
una actitud poco ética si tenemos la certeza
que el trazado paroxístico y el trastorno del
lenguaje no tienen ninguna relación, pero no
menos grave sería el hecho de no tratar una epilepsia
con la medicación adecuada. El estudio de un
numero suficiente de casos con una metodología
rigurosa constituye el único modo de poder llegar
a responder a una pregunta trascendente por sus implicaciones:
?Puede la epilepsia manifestarse como un trastorno puro
de la comunicación y la cognición?.
TRASTORNOS
ESPECÍFICOS DEL DESARROLLO DEL LENGUAJE
El
hecho de que en la práctica diaria se llegue
al diagnóstico de Disfasia o trastorno específico
del desarrollo del lenguaje por exclusión de
todas las posibles causas, tanto patológicas
como socioculturales que pueden interferir en la adquisición
del lenguaje, nos da idea del gran desconocimiento que
seguimos teniendo sobre el tema a pesar de todos los
avances en investigación fundamentalmente por
técnicas de neuroimagen funcional.
Cuando
un niño llega a la consulta con retraso en la
adquisición del lenguaje y hay que determinar
si se trata de una trastorno específico debemos
descartar aquellos factores de tipo emocional, conductual
o situacional que están influyendo. Igualmente,
hay que descartar la existencia de una patología
definida. Para ello es necesaria una exploración
cuidadosa neurológica y psicológica. La
valoración de todos los datos nos indicará
si es necesario llevar a cabo estudios médicos
complementarios. En caso de no necesitarse, habrá
que definir las orientaciones y las prácticas
terapéuticas adecuadas. Las pautas de actuación
general son las siguientes:
a)- Historia clínica: Incluye los datos del entorno
sociocultural y la escuela. Dentro de la historia clínica,
algunos de los informes ineludibles son los relacionados
con los siguientes aspectos: embarazo, parto, periodo
neonatal, hitos madurativos (motores y lenguaje), enfermedades,
hábitos de autocuidado, hiperactividad y déficit
de atención, motricidad gruesa y fina, cambios
de conducta, antecedentes familiares; y de una manera
especial, la entrada en la escuela, las dificultades
de aprendizaje en general y de la lectura en especial.
b)- Exploración neurológica: Incluye la
valoración de los pares craneales, vías
piramidales, extrapiramidales y cerebelosa, fuerza y
tono muscular y fenotipo, así como exploración
somática general. De la normalidad de estos datos
depende la confirmación diagnóstica de
trastorno específico del aprendizaje. En caso
de encontrar datos patológicos estaríamos
ante un trastorno secundario a una causa, es decir la
dislexia sería consecuencia de la patología
de base hallada.
c)- Exploración neuropsicológica: Incluye
la valoración del cociente de inteligencia, el
desarrollo del lenguaje espontáneo (fluidez,
vocabulario, construcción de la sintaxis), la
memoria, la atención, la coordinación
visomotora. Para esta exploración deben ser utilizadas
pruebas estandarizadas y observaciones conductuales
siguiendo protocolos y pautas. Esto nos da una valoración
objetiva de la situación del paciente y nos permite
la comparación con otros pacientes y consigo
mismo a la hora de valorar la evolución.
d)- Exámenes complementarios: Este es un tema
tremendamente controvertido. La utilización de
los exámenes médico- complementarios en
los trastornos específicos del lenguaje y aprendizaje
ha sido desaconsejado durante mucho tiempo. Sin embargo,
si debemos llegar a un diagnóstico por exclusión,
deberemos utilizar los medios que poseemos para asegurarnos
que realmente la normalidad del niño es absoluta.
La posibilidad de estar frente a una premutación
del cromosoma x frágil o el creciente interés
de las investigaciones que ponen de manifiesto la interferencia
de descargas paroxísticas epileptiformes, aún
en ausencia de crisis clínicas, sobre el aprendizaje
hacen que la negativa a efectuar exámenes complementarios
deba ser reconsiderada.
Frente
a un niño que no adquiere el lenguaje, es difícil
asegurar que nos encontramos ante una disfasia sin causa
patológica subyacente si no tenemos la certeza
de normalidad al menos en las tres siguientes pruebas
las cuales deberían constituir el mínimo
de exámenes aconsejables: 1) estudio de ADN para
descartar síndrome x frágil; 2) RM cerebral
para descartar ectopias o malformaciones y c) EEG de
vigilia y sueño para descartar descargas paroxísticas,
aún en ausencia de crisis clínicas.
La
normalidad de los resultados, nos llevará al
diagnóstico de trastorno específico del
lenguaje. En este punto, la actuación médica
remite y será el apoyo pedagógico específico
el que guíe la actitud terapéutica, con
el soporte psicológico y logopédico. El
hallazgo de una patología neurológica
concreta no implica que se deba descuidar lo apuntado
en el punto anterior. A ello habrá que añadirse
el tratamiento médico específico de la
enfermedad de que se trate.
Ahora
bien, el diagnóstico de TEDL puede ser tarea
difícil y en algunos casos el trastorno de la
comunicación que producen es de tal entidad que
implique un diagnóstico diferencial con el espectro
autista.
Recordemos
que en condiciones normales la fonología ha terminado
su maduración a los 4 años y la morfosintaxis
a los 6. Estudios por resonancia magnética funcional
muestran que el lenguaje se encuentra definitivamente
lateralizado a los 7 años (52). El diagnóstico
sindrómico del tipo de trastorno ayuda a comprender
la clínica y la evolución. Sigue siendo
una de las clasificaciones de mayor utilidad la ofrecida
por Rapin y Allen (53) y que divide los TDEL en tres
grandes grupos, según afecten a la expresión,
a comprensión y expresión o al procesamiento.
Sus características se detallan en la tabla 2.
De
todos ellos, el mas frecuente es el trastorno mixto
fonológico- morfosintáctico, y el mas
severo la agnosia auditiva verbal(AAV). Es este último
el que va a plantear el diagnóstico diferencial
con el autismo. La AAV conoce como dato guía
una severísima alteración de la comprensión
del lenguaje, que en su grado sumo incluye a los sonidos
no verbales de la vida cotidiana (puerta, campana, coche,
agua….etc..etc). Esta ausencia de comprensión
repercute en la expresión, es decir, como consecuencia
del no entender, el niño no adquiere el lenguaje
expresivo. Como consecuencia también de la falta
de comprensión el niño tiende a aislarse,
pierde el interés por comunicar al ser consciente
de su trastorno, puede adoptar conductas oposicionistas
y/o hipercinéticas y hasta llegar al diagnóstico
de disfasia, estos pequeños pasan por sordos,
retrasados mentales o autistas y es necesaria una cuidadosa
evaluación neuropsicológica para establecer
el correcto diagnóstico de TEDL, en el que son
claves e Inventario De Espectro Autista (54) compuesto
por doce dimensiones (y cuatro niveles de afectación)
agrupadas en cuatro escalas: Desarrollo social, Comunicación
y Lenguaje, Anticipación y Flexibilidad, Simbolización;
y la escala Leiter (55) para valorar el cociente intelectivo.
Las
causas últimas de la disfasia son desconocidas,
aunque alteraciones en función metabólica
cerebral áreas del lenguaje, disfunciones cerebelosas
y alteraciones de la migración neuronal han sido
publicadas (56- 62). .
El
pronóstico es incierto, no existiendo en la actualidad
marcadores biológicos que nos permitan predecir
la normalización y grado de normalidad de la
alte4ración que presenta el paciente. Es fundamental
la concienciación del entorno frente a las necesidades
específicas de estos niños gran número
de los cuales presentaran posteriormente dificultades
en el aprendizaje del lenguaje escrito, siendo el binomio
disfasia- dislexia una situación a prever.
EN
CONCLUSIÓN: El trastorno específico
del lenguaje no debe ser considerado una enfermedad
pero si que tenemos la evidencia que existe un funcionamiento
distinto, que no patológico, del cerebro de estos
niños y en definitiva, un niño disfásico
no es un enfermo, es una persona inteligente, cuyo cerebro
posee una capacidad distinta a la norma de procesar
la información y cuyo futuro depende del conocimiento
y adecuado tratamiento de esta circunstancia lo que
implica una seria responsabilidad del entorno familiar,
social, médico y por encima de todo ello, del
entorno escolar.
TABLA
2- CLASIFICACIÓN DE RAPÍN Y ALLEN- TEDL
TRASTORNOS
EXPRESIVOS
DISPRAXIA
VERBAL: Alteración severa de la fluencia verbal.
Articulación
de la palabra muy alterada.
Comprensión
preservada.
DÉFICIT
DE PROGRAMACIÓN FONOLÓGICA:
Lenguaje
fluido pero de difícilmente inteligible.
Comprensión
preservada.
TRASTORNOS
MIXTOS (EXPRESIÓN Y COMPRENSIÓN)
DÉFICIT
FONOLÓGICO- SINTÁCTICO:
Fluencia
alterada.
Articulación
de la palabra, alterada.
Alteración
de la estructura gramatical de la frase.
Alteración
en grado diverso de la comprensión, pero siempre
de mejor calidad que la expresión.
AGNOSIA
AUDITIVA:
Alteración
severa de la fluencia verbal.
Articulación
de la palabra muy alterada.
Expresión
limitada a pequeñas frases o palabras aisladas.
Comprensión
del lenguaje severamente alterada e incluso abolida.
ALTERACIÓN
DEL TRATAMIENTO CENTRAL Y PROCESAMIENTO DEL LENGUAJE
DÉFICIT
SEMÁNTICO- PRAGMÁTICO:
Fluencia
exagerada; logorrea.
Articulación
de la palabra, normal.
Estructura
de la frase, normal.
Cambio
contínuo de tema conversacional (conversador
de salón o "papagayo"), sin respetar
el turno de conversación.
Frases
estereotipadas.
Comprensión
alterada frente a enunciados complejos.
DÉFICIT
LÉXICO- SINTÁCTICO:
Palabra
fluente.
Pseudotartamudez
por dificultad en la evocación de la palabra.
Articulación
normal.
Sintaxis
inmadura.
Dificultad
para relatos y formulaciones del lenguaje complejas.
Comprensión
alterada de enunciados complejos.
BIBLIOGRAFÍA
1-
Marín-Padilla, M. (1995) Desarrollo de la neocorteza
cerebral humana. Revista de Neurología Clínica,
23 (Supl.3): S261- S268.
2-
Narbona, J. y Fernández, S. (1996) Fondements
neurobiologiques du développement du langage.
En Le langage de l?enfant. Aspects normaux et pathologiques,
(ed Masson) Paris.
3-
De Otero Y, (1998) Síndrome x frágil y
discapacidad mental hereditaria.
Ministerio
de educación y Consumo.
4-
Artigas Pallarés, J; Brun, C; Gabau, E. (2001)
Aspectos médicos y psicológicos del síndrome
x frágil. Revista de Neurología Clínica,
2 (1): 42- 54.
5-
Aicardi J. Epilepsy: The hidden part of the iceberg.
Eur J Paediatr Neurol 1999; 3: 197- 200.
6- Bulteau C, Jambaque I, Viguier D, Kieffer V, Dellatollas
G, Dulac O. Epileptic syndromes, cognitive assessment
and school placement: A study of 251 children. Dev Med
Child Neurol 2000; 42: 319- 327.
7-
Rugland AL. Neuropsychological assesement of cognitive
functioning in children with epilepsy. Epilepsia
1990;
31(Suppl.4):S41-S44.
8-
Schoenfeld J, Seindenberg M, Woodard A, Hecox K, Inglese
C, Mack K, et al. Neuropsychological and behavioral
status of children with complex partial seizures. Dev
Med Child Neurol 1999; 41: 724-731.
9-
Stores G. Electroencephalographic parameters in assesing
the cognitive function of children with epilepsy. Epilepsia
1990; 31(Suppl.4): S54-S49.
10-
Landau WM, Kleffner FR. Syndrome of acquired aphasia
with convulsive disorder in children. Neurology 1957;
7( 8): 523- 530.
11-
Hirsch E, Marescaux C, Maquet P, Metz- Lutz MN, Kiesmann
M, Salmon E, et al. Landau- Kleffner syndrome: A clinical
and eeg study of five cases. Epilepsia 1990; 31 (6):
756- 767.
12-
Deona T, Beaumanoir a, Gaillard F, Assal G. Acquired
aphasia in childhood with seizure disorder: A heterogeneus
syndrome.
Neuropädiatrie 1977; 8(3): 263- 273.
13-
Deona T, Fletcher P, Voumard C. Temporary regresion
during language acquisition: A linguistic analysis of
a 2 ? -year-old child with epileptic aphasia. Dev Med
Child Neurol 1982; 24: 156-163.
14-
Tassinari CA, Bureau M, Dravet C, Dalla Bernardina,
B, Roger J. Epilepsie avec pointes- Ondes continues
pendant le sommeil lent. En Roger J, Dravet C, Bureau
M, Dreiffus FE, Wolf P, eds. Les syndrômes epiléptiques
de l?enfant et de l?adolescent. London/Paris: John Libbey,
1984. p.198- 209.
15-
Aicardi J, Chevrie JJ.Atypical benign partial
epilepsy of childhood. Dev Med Child Neurol 1982; 24:
281- 292.
16-
Nieto Barrera M, Aguilar Quero F, Montes E, Candau R,
Prieto P. Síndromes epilépticos que cursan
con complejos punta- onda continuos durante el sueño
lento.Rev Neurol 1997; 25 (143): 1045- 1051.
17-
Veggiotti P, Beccaria F, Guerrini R, Capovilla G, Lanzi
G. Continuous spike and wave activity during slow wave
sleep: Syndrome or EEG pattern?. Epilepsia 1999; 40:
1593-1601.
18-
Staden U, Isaacs E, Boyd SG, Brandl U, Neville BGR.
Language dysfunction in children with rolandic epilepsy.
Neuropediatrics 1998; 29: 242- 248.
19-
Croona C, Kihlgren M, Lundberg S, Eeg- Olofsson O, Eeg-
Olofsson K. Neuropsychological findings in children
with benign childhood epilepsy with centrotemporal spikes.
Dev Med Child Neurol 1999; 41: 813-818.
20-
Tuchman RF, Rapin I, Sholmo S. Autistic and dysphasic
children. II: Epilepsy. Pediatrics 1991; 88(6): 1211-
25.
21-
Filipek PA, Accardo PJ, Baranek GT, Cook EH, Dawson
G, Gordon B, et al. The secreening and diagnosis of
autistic spectrum disorders. J Autism Dev Disord 1999;
29 (6): 439- 484.
22-
Herranz JL. Efectos neuropsicológicos de los
fármacos antiepilépticos. Rev Neurol 1997;
25(Supl 4): S433- S438.
23-
Pestana EM, Sardinas N, Trujillo C. Factores que deben
considerarse en la valoración del rendimiento
intelectual en el niño epiléptico. Rev
Neurol 1997; 25 (144): 1225- 1228.
24-
Kasteleinj-Nolst Trenité DGA, Rimersa JBJ, Binnie
CD, Smith AM, Meinardi H. The influence of subclinical
epileptiform EEG Discharges on driving behaviour. Electroenceph
Clin Neurophysiol 1987; 67:167-170.
25-
Kasteleinj-Nolst Trenité DGA, Siebelink BM, Berends
SGC, van Strien JW, Meinardi H. Lateralized effects
of subclinical epileptiform EEG discharges on scholastic
performance in children. Epilepsia 1990; 31(6): 740-
746.
26-
Byring RG, Salmi TK, Sainio KO, Örn HP. EEG in
children with spelling disabilities. Electroenceph Clin
Neurophysiol 1991; 79: 247-255.
27-
Binnie CD, Marston D. Cognitive correlates of interictal
discharges. Epilepsia 1992; 33 (Suppl 6): S11- S17.
28-
Binnie CD. Significance and manegement of transitory
cognitive impairment due to subclinical EEG discharges
in children. Brain Develop 1993; 15(1): 23- 30.
29-
Tuchman RF., Rapin I. Regresion in pervasive developmental
disorders: Seizures and epileptiform electroencephalogram
correlates. Pediatrics 1997; 99 (4): 560-566.
30
- Sato S, Dreiffus FE. Electroencephalographic findings
in a patient with developmental expresive aphasia. Neurology
1973; 23: 181-185.
31-
Maccario M, Hefferen S, Keblusek K, Lipinski K. Developmental
dysphasia and electroencephalographic abnormalities.
Dev.Med.Child Neurol 1982;24: 141-155.
32-
Echenne B. Dysphasies et épilepsie. ANAE 1990;
3: 138- 143.
33-
Echenne B, Cheminal R, Rivier F, Negre C, Touchon J,
Billiard M. Epileptic abnormalities and developmental
dysphasias: A study of 32 patients. Brain Dev 1992;
14: 216- 225.
33-
Picard A, Cheliout-Heraut F, Bouskraoui M, Lemoine M,
Lacert P, Delattre J. Sleep EEG and developmental dysphasia.
Dev Med Child Neurol 1998; 40: 595-99.
34-
Chevrie- Muller C, Narbona J. Classification des troubles
du langage observés dans l?enfance. En Chevrie-
Muller C, Narbona J, eds. Le langage de l?enfant: Aspects
normaux et pathologiques. Paris/Milan/ Barcelona.Ed.
Masson; 1996. p. 195- 199.
35-
Beaumanoir A. Le syndrome de Landau- Kleffner. En Roger
J, Dravet C, Bureau M, Dreiffus FE, Wolf P, eds. Les
syndrômes epiléptiques de l?enfant et de
l?adolescent. London/Paris: John Libbey, 1984. p.185-
195.
36-
Mantovani JF, Landau WM. Acquired aphasia with convulsive
disorder: Course and prognosis. Neurology 1980; 30:
524- 529.
37-
Gordon N. Acquired aphasia in childhood: The Landau-
Kleffner syndrome. Dev Med Child Neurol 1990; 32: 267-
274.
38-
Deona T, Peter C, Ziegler AL. Adult follow-up of the
acquired aphasia- epilepsy Syndrome in childhood. Report
of 7 cases.
Neuropediatrics
1988; 20: 132- 138.
39-
Mantovani JF. Autistic regression and Landau- Kleffner
syndrome: progress or confusion?. Dev Med Child Neurol
2000; 42: 349- 353.
40-
Rapin I. Understanding childhood language disorders.
Pediatrics 1988; 10: 561- 566.
41-
Rapin I. Developmental Language Disorders: A Clinical
Update. JChild Psychol.Psychiat 1996; 37(6): 643-55.
43-
Duvelleroy- Hommet C, Billard C, Lucas B, Gillet P,
Barthez MA, Santinni JJ, et al. Sleep and developmental
dysphasia: Lack of a consistent relationship with paroxysmal
eeg activity during sleep. Neurology 1995; 26: 14- 18.
44-
Bax M. Problems for the child with epilepsy. Dev Med
Child Neurol 1999; 41: 723.
45-
Mantovani J. Treat the patient, not the EEG?. Dev Med
Child Neurol 2000; 42: 579.
46-
Deona T, Zesiger P, Davidoff V, Maeder M, Roulet E.
Benign partial epilepsy of childhood: A longitudinal
neuropsychological end eeg study of cognitive fonction.
Dev Med Child Neurol 2000; 42: 595- 603.
47-
Narbona J, Chevrie- Muller C. Évaluation neuropsychologique.
En Chevrie- Muller C, Narbona J, eds. Le langage de
l?enfant: Aspects normaux et pathologiques. Paris/Milan/
Barcelona.Ed. Masson; 1996. p. 109- 130.
48-
Dalla Piazza S. L?evaluation en neuropsychologie de
l?enfant. Neuropsychiatr Enfance Adolesc 1997; 45(1-2):
6- 22.
49-
Soprano AM, García EF, Caraballo R, Féjerman
N. Acquired epileptic aphasia: Neuropsychologic follow-up
of 12 patients. Pediatr Neurol 1994; 11(3): 230-235.
50-
Wasterlain CG, Shirasaka Y. Seizures, brain damage and
brain development. Brain Dev 1994; 16: 279-95.
51-
Aicardi J. La epilepsia como un trastorno no paroxístico.
Acta Neuropediatr 1996; 2: 248- 257.
52-
Sans A, y otros (2001).Resonancia magnética funcional:
su utilidad en neuropsicología
Infantil.
Rev Neurol ,2 (1), 72- 85.
53-
Rapin I, (1982) Developmental languaje disorders.
En
Rapin I. (Ed), Children with brain dysfunction. Raven
Press. New York.
54-
Riviere A, (1997) Tratamiento y definición del
espectro autista.
En
Riviere A, Martos J, El tratamiento del Autismo. Nuevas
perspectivas. Madrid. IMSERSO- APNA.
55-Leiter
RG. Leiter International Performance Scale. USA: Stoelting
Co.; 1980.
56-
Habib, M., Robinchon F, Démonet JF. (1996) El
singular cerebro de los disléxicos. Mundo Científico,
172: 848- 853.
57-
Nicolson, R.I. y otros (1999) Association of abnormal
cerebellar activation with motor learning difficulties
in dyslexic adults. Lancet, 353: 1662- 1667.
58-
Potter, N.T. y Tarleton J. (1998) Neurogenetics in developmental
and behavioural pediatrics: Advances in molecular diagnosis.
Developmental and behavioural pediatrics, 19:117- 130.
59-
Rae C. y otros (1998) Metabolic abnormalities in developmental
dyslexia detected by Hmagnetic Resonance Spectroscopy.
Lancet, 351: 1849- 1852.
60-
Rumsey, M. y otros (1992) Failure to activate the left
temporoparietal cortex in dyslexia. An oxygen 15 positron
emission tomografic study. Archives of Neurology, 49:
527- 534.
61-
Schmahmann, J.D. (1991) An emergin concept. The cerebellar
contribution to higher function. Archives of Neurology,
48: 1178- 1187.
62-
Shaywitz, S. y otros (1998) Functional disruption in
the organization of the brain for reading in dyslexia.
Proceedings of the national academic for science USA
95, 2636-41.